Mercator
Multiple Whole-Genome Orthology Map Construction
Colin Dewey,
Lior Pachter
[Introduction]
[Methods]
[Maps/Alignments]
[Source Code]
[References]
Whole-genome homology maps attempt to identify the evolutionary
relationships between and within multiple genomes. The term
"syntenic" is often used to describe regions of multiple genomes that
are believed to have evolved from the same region in an ancestral
genome. However, it has been pointed out that this use of the term is
incorrect (Passarge et al. 1999) and thus we
will use the terms "homologous", "orthologous", and "paralogous"
instead. Ideally, given K genomes, we would like to identify all
orthologous genomic regions as well as paralogous regions within each
genome and hypothetical ancestral genome. Maps listing these
relationships are extremely valuable to researchers performing
comparative analyses of genomic sequence. Here we present our initial
work in the form a program called Mercator that constructs
orthology maps between multiple whole genomes.
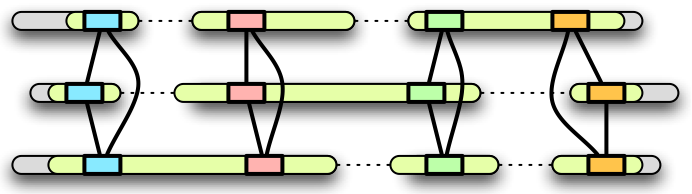
Our basic strategy in building homology maps is to use exons that are
orthologous in multiple genomes as map "anchors." Given K genomes,
the steps in the map construction are as follows:
- For each genome, obtain a set of exon annotations. These
annotations can be a combination of both exon predictions (e.g. Genscan)
and annotations that have been experimentally verified (e.g. RefSeq).
Ideally, we would like to have these annotations be as sensitive as
possible. Specificity is not a concern, as incorrect annotations are not
likely not have significant alignments with other gene annotations.
- Compare all exons against all exons in other genomes and record
significant alignments between exons. Currently, we use BLAT to do this all-vs-all comparison with
alignments being performed in protein space.
- Construct a graph with each vertex corresponding to a exon and
edges between vertices whose corresponding exons have significant
alignments.
- Identify cliques in this graph. These cliques are potential
anchors to be used in the map.
- Starting with the largest cliques (those that have exons in all
or most of the genomes), join neighboring (adjacent in genomic
coordinates, in each genome) cliques to form runs.
Smaller cliques that are inconsistent with runs formed by larger
cliques are filtered out. After the smallest cliques have been
considered, cliques that are not part of a run are discarded.
- The extents of each run in each genome are outputted as
orthologous segments. The cliques from each run are used to output
the exact genomic coordinates of anchors within each orthologous
segment. These anchors can be used by genomic alignment programs
(such as MAVID) to do a detailed alignment of
each orthologous segment.
Mercator is free software under the
GNU General Public
License and is available as part of Colin Dewey's
source
code distribution.
Binaries may be made available upon request.
-
S. Lall, D. Grün, A. Krek, K. Chen, Y. Wang, C. Dewey, P. Sood, T. Colombo, N. Bray, P. MacMenamin, H. Kao, K. Gunsalus, L. Pachter, F. Piano, and N. Rajewsky.
A genome-wide map of conserved microRNA targets in C. elegans.
Current Biology. (2006), In press.
-
Bray, N and Pachter, L. (2003)
MAVID: Constrained Ancestral Alignment of Multiple Sequences.
Genome Research,
14:693-699.
-
Kent, W.J. (2002)
BLAT:
The BLAST-like alignment tool.
Genome Research,
12: 656-664.
-
Passarge, E., Horsthemke, B., and Farber, R.A. (1999)
Incorrect use of the term synteny.
Nature Genetics, 23:387
-
Rat Genome Sequencing Project Consortium. (2004)
Genome sequence of the Brown Norway rat yields insights into mammalian evolution.
Nature vol. 428, 493-521.